GENERAL INFORMATION
Extraskeletal Ewing Sarcoma (EES) is a malignant small round-blue cell tumor than arises most often in Children and adolescents. Tefft et al in 1969 described the first series of Extraskeletal Ewing Sarcoma that arose from the paravertebral soft tissues. Ewing sarcoma, although a rare type of sarcoma, is most typically a type of sarcoma that arises from bone. However more rarely Ewing Sarcoma can arise from the soft tissues and is then termed Extraskeletal Ewing Sarcoma. EES is a rare tumor composed of monotonous primitive small round blue-celsl distributed in pieces or lobules. It’s has an annual incidence rate of 1-3 cases per million. Ewing sarcoma and primitive neuroectodermal (PNET) tumor fall in the same family of tumors.
CLINICAL DATA
High grade sarcoma
•
Rare soft tissue tumor, indistinguishable from
Ewing sarcoma of bone
•
Usually affects patients age 30 or less, occasionally age >50
•
Slight male preference
•
Most commonly metastasizes to lung and bones
DIFFERENTIAL DIAGNOSIS
•
Lymphoma
•
Rhabdomyosarcoma
•
Infection/abscess
CLINICAL PRESENTATION
Sign/Symptoms
•
Usually a large and deep mass
•
Rarely subcutaneous
•
Rapidly growing
•
Usually painless (pain in 30% of cases)
Prevalence
•
Much less common than skeletal Ewing sarcoma
•
Slight predilection for males
•
Rarely effects afro-americans
•
Occurs in all ages but usually younger population
•
Preference for individuals between 15 and 40 years
Site
•
Commonly in soft tissue of lower extremities and thorax
RADIOGRAPHIC PRESENTATION
Plain x-ray
•
No specific radiological features
•
May reveal a soft tissue mass
•
No mineralization
•
If adjacent to a bone may show underlying bony erosion
CT
•
Shows attenuation similar to the muscle
•
May be heterogenous with areas of necrosis and hemorrhage
MRI
•
Low to intermediate signal intensity on T1W
•
High signal intensity on T2W
•
Usually heterogeneous from necrosis and hemorrhage
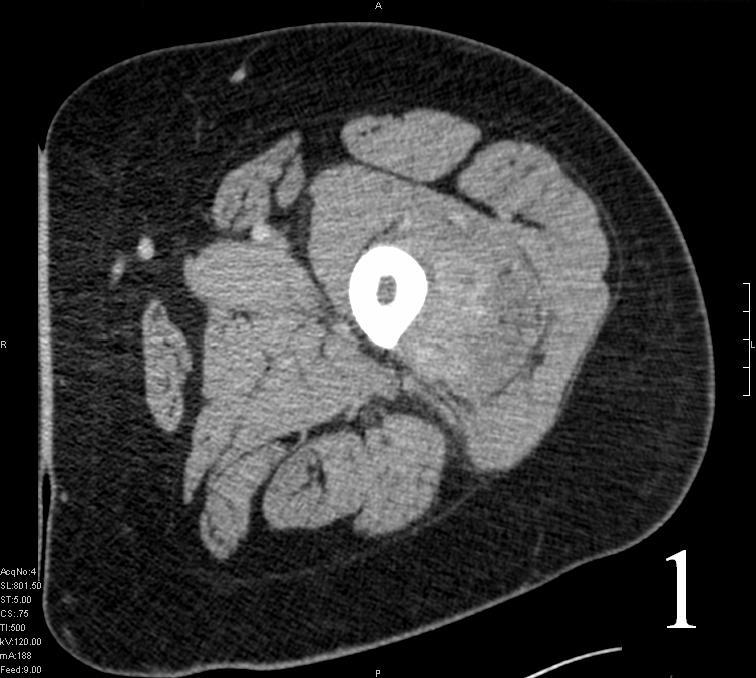
Fig. 1
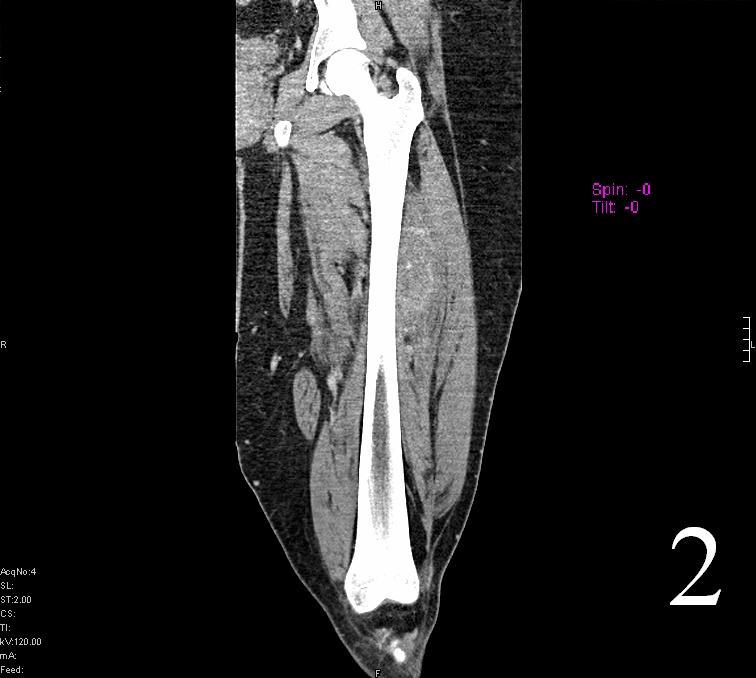 Fig. 2
Fig. 1-2 Axial (Fig. 1) and coronal CT (Fig. 2) reconstruction of the femur and thigh shows an Extraskeletal Ewing Sarcoma arising in the soft tissues adjacent to the femur
Fig. 2
Fig. 1-2 Axial (Fig. 1) and coronal CT (Fig. 2) reconstruction of the femur and thigh shows an Extraskeletal Ewing Sarcoma arising in the soft tissues adjacent to the femur
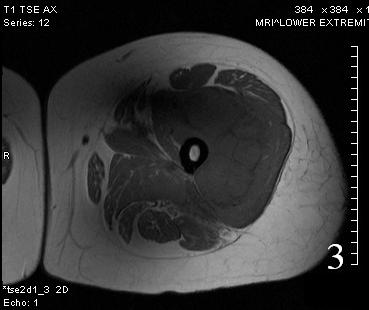 Fig. 3
Fig. 3
 Fig. 4
Fig. 4
Fig. 3-4 Axial (Fig. 3) and coronal (Fig. 4) T1 W MRI of the thigh shows an Extraskeletal Ewing Sarcoma that is iso-intense with the adjacent musculature.
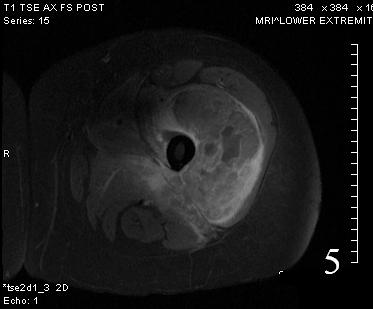 Fig. 5
Fig. 5
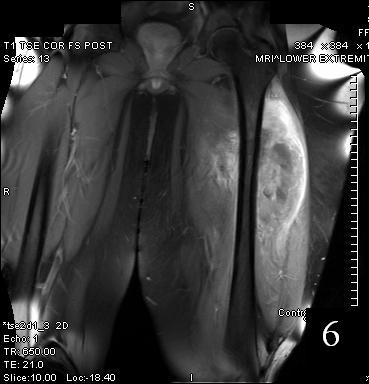 Fig. 6
Fig. 6
Fig. 5-6 Axial (Fig. 5) and coronal (Fig. 6)T1 post contrast MRI shows a heterogenous mass with enhancement post contrast
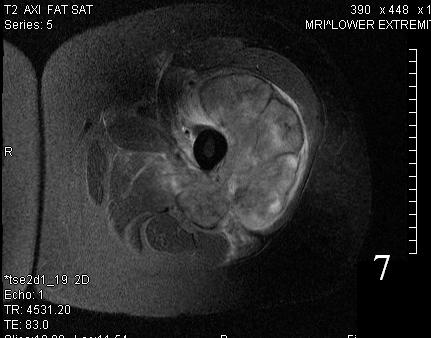 Fig. 7
Fig. 7
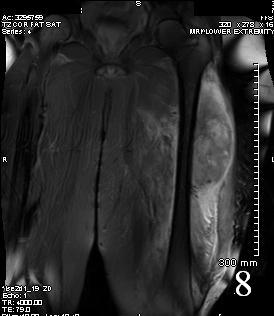 Fig. 8
Fig. 7-8 Axial (Fig. 7) and coronal (Fig. 8) T2W shows heterogenous high signal intensity
Fig. 8
Fig. 7-8 Axial (Fig. 7) and coronal (Fig. 8) T2W shows heterogenous high signal intensity
PATHOLOGY
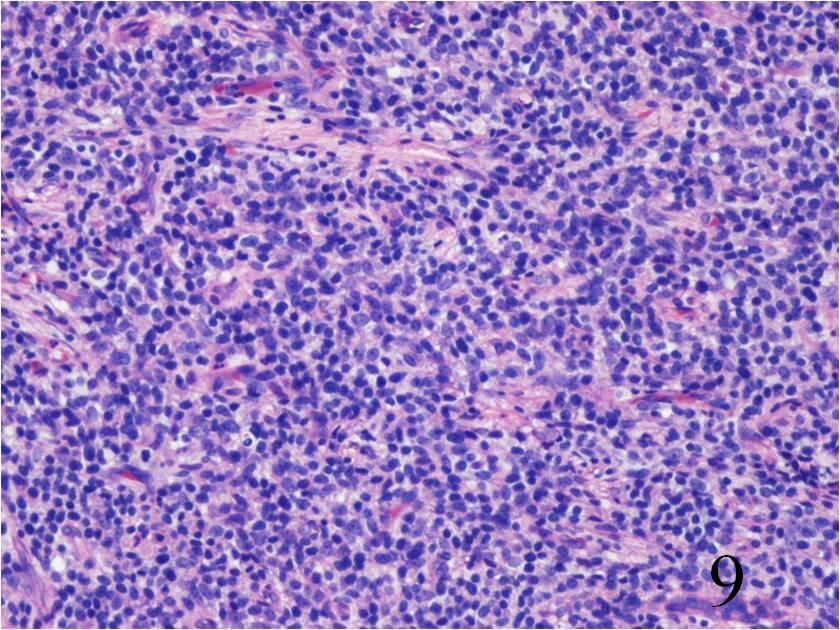 Fig. 9
Fig. 9
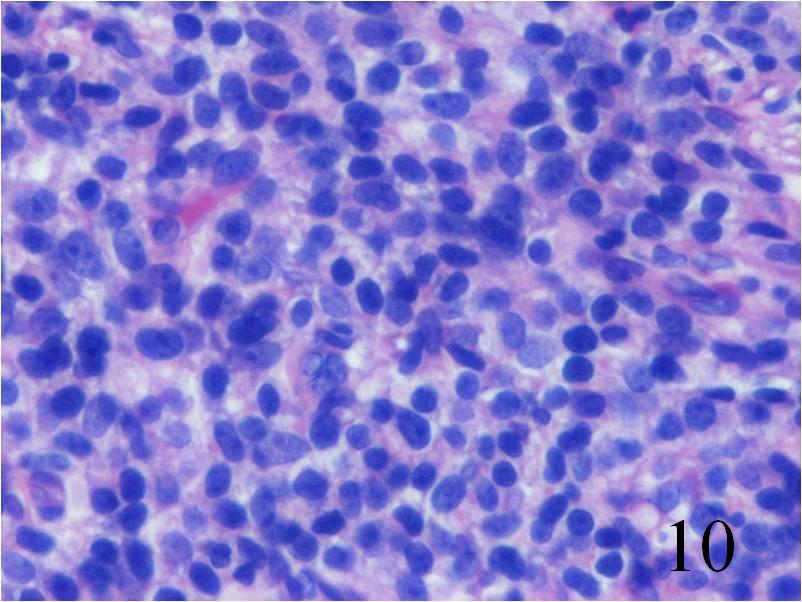 Fig. 10
Fig. 10
Fig. 9-10 Microscopic: Low power (Fig. 9) and high power (Fig. 10) views of an Extraskeletal Ewing Sarcoma shows a monotonous Collectionb of small round blue cells. The cells have large nuclei and minimal cytoplasm.
IMMUNOHISTOCHEMISTRY
Positive
o
CD 99
o
PSA
o
Vimentin
•
Negative
o
S-100
o
NF
o
CK 7
o
CK 19
o
Leukocyte antigen
PROGNOSIS
Biological Behavior
Tumor cells often express MIC2 gene
o
Demonstrate several novel reciprocal chromosomal translocations and fusion gene transcripts
o
t(11;22) chromosomal translocation most commonly encountered and often aides in diagnosis
•
High rate of metastasis
o
Most commonly in the lungs and bones
•
Best prognosis in patients age<16 years who underwent surgical resection and chemotherapy.
•
Patients with metastatic disease generally die within 2 years
•
50% 5-years survival rate in patients who present with localized disease
TREATMENT
Complete excision; Limb Sparing Surgery
•
Radiotherapy may be indicated
•
Multi-drug chemotherapy (cyclophospphamide, ifosfamide, etoposide, doxorubicin and vincristine)
